10 Release Notes
10.1 2025A
Pseudopotential Update
LDA and PBE pseudopotentials for elements of periods 4-6 (K Ca Sc Ti V Fe Co Ni Cu Zn Ga Ge As Se Br Sr Y Zr Nb Mo Tc Ru Rh Pd Ag Cd In Sn Sb Te Hf Ta W Re Os Ir Pt Au Hg Pb Bi Po) have been updated to version 1.1, improving computational accuracy and lowering the cutoff energy.
Functional Improvements
Optimized memory consumption during pcharge calculation
Comprehensive code optimization significantly improves computation speed.
10.2 Release Notes for Version 2023A
New Features
Support for constant potential method in SCF calculations
Implicit solvent model is supported.
AIMD calculations now include a Langevin thermostat (barostat), adding support for NPT/NPH ensembles; aimd.thermostat=none is renamed to SA (simulated annealing).
Support for fitting and band interpolation calculations with Maximally Localized Wannier Functions (MLWF).
Pseudopotential Updates
LDA and PBE pseudopotentials for the first three periods (H, He, Li, Be, B, C, N, O, F, Ne, Na, Mg, Al, Si, P, S, Cl, Ar) are released in version 1.1, improving computational accuracy and reducing the cutoff energy.
IO Tuning
Added HDF5 format files as the default output file format for DS-PAW, and the JSON format output files will no longer be maintained.
Modified the output for the DS-PAW.log parameters.
Remove tmp folder
Feature Optimization
Added Pulay option for scf.mixType
Added ‘atom’ and ‘shape’ options to relax.freedom
Added the relax.pressure parameter.
Added parameters related to FFT grid: cal.FFTGrid, cal.supGrid
Added support for the cal.opticalGrid parameter when io.optical=true.
Added support for alternative writing styles in band.kpointsNumber
Added the corr.dftuForm parameter to determine the DFT+U method type.
Added support for exchange-correlation functionals compatible with semi-empirical VDW corrections.
Updated 2023A 2024/04/03
Supplement the output of the wave function derivative with respect to k during optical calculations.
Feature optimization
Added sys.spinDiff parameter to restrict the difference in the number of spin-up and spin-down electrons.
Added corr.coreEnergy parameter to control whether core electron energy levels are calculated.
Fixed a bug where the mag parameter was read incorrectly in some cases.
Updated on 2024/03/15 (2023A)
Feature Optimization
Fixed the issue where single-atom calculations of H, using PWSP pseudopotentials, resulted in errors.
Optimized the HSE calculation code to avoid Intel errors.
2023A Updated on 2024/01/12
IO adjustments
- Added Fermi level information to rho.bin.
When task=dos/band, the Fermi level can be directly read from rho.bin without reading from system.json; (This version is compatible with older versions of rho.bin that do not contain EFermi, but the older version of DS-PAW cannot read the rho.bin output by this version)
Added parameter band.EfShift, which controls whether to read EFermi from rho.bin when task=band is used.
Supplement the ##PARAMETERS## section of DS-PAW.log with io.band and io.dos output.
Added band information output to DS-PAW.log
Feature optimization
Fixed an issue where the partial results were incorrect when task=pcharge;
Added parameter scf.timeStep to adjust the convergence of electronic steps when cal.methods is set to 4 or 5.
- Added parameter task=optical.
Renamed the parameter cal.opticalGrid to optical.grid
Added parameters optical.KKEta, optical.smearing, optical.sigma, optical.Emax
2023A Updated on 2023/10/07
IO adjustments
Fixed an issue where a warning would be incorrectly output when sys.functional was not defined in the input file when sys.hybrid = true.
Fixed an issue where the number of FinalStep outputs did not match the number of step-XX outputs when task=aimd/relax.
Feature optimization
Optimized the acoustic calculation module to strictly adhere to the acoustic sum rule.
Fix the issue of abnormal forces in the complex density functional task=relax.
2023A Updated on 2023/6/21
IO Adjustment
Add force-related outputs in neb0X.h5 during NEB calculations.
Change E2095 error to warning and add relevant explanations
Adjust the output format for task=wannier
Increase the number of significant digits for reading and writing sys.fixedP-related data and parameter passing.
Feature Optimization
Optimized the band.unfolding algorithm to reduce the probability of memory crashes.
In the FixedPotential iteration, precision control is implemented for the output .input.json file to prevent redundant calculations in some cases.
Fixed data format issues in some .txt output files, preventing data output stacking.
2023A Update: May 9, 2023
I/O Adjustments
Adjusted output information related to task=neb, sys.hybrid=true, and scf.mixType=Broyden.
Fixed the error where HeatCapacity output was null when task=phonon and phonon.thermal=true.
Feature Enhancements
Added fixed lattice and atom position information to latestStructure.as; fixed a bug related to incorrect mag information output.
Adjusted the io.magProject default value to true when sys.spin is set to collinear/non-collinear.
Fixed an issue with incorrect reading of mag-related information from relax.json/relax.h5 during continued calculations.
10.3 Function Summary
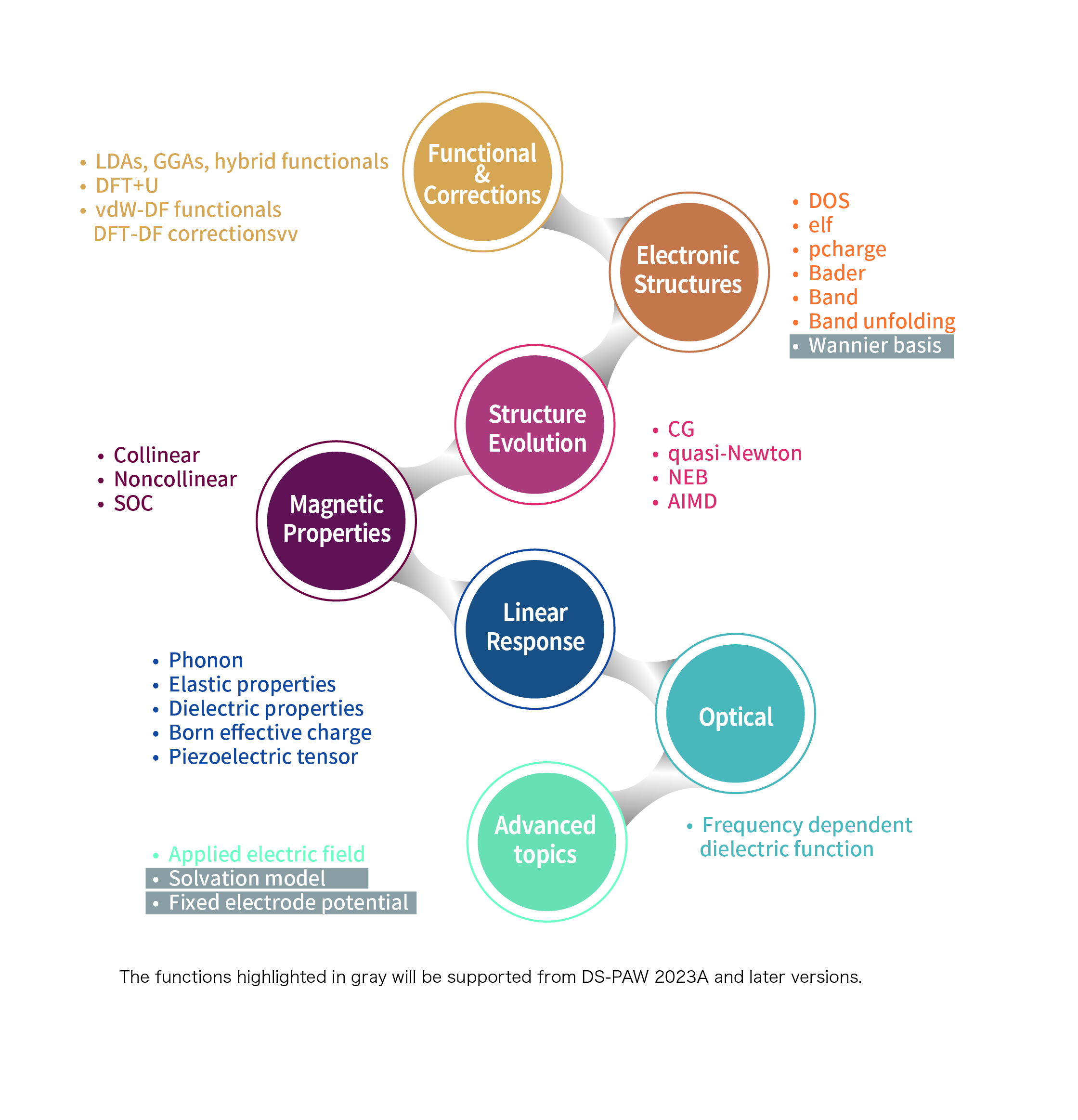
10.4 Release History
2022A
New Features
Support for revPBE/PBEsol/RPBE exchange-correlation functionals
Supports vdW functionals: vdW-optPBE, vdW-optB88, vdW-optB86b, vdW-DF, vdW-DF2, and vdW-revDF2
Supports simulation of external electric field effects
Supports NEB calculations with variable lattice systems (solid state NEB, ssNEB)
Supports calculation of ferroelectric polarization using modern polarization theory
Support band unfolding functionality
Support calculation of Helmholtz free energy/constant volume heat capacity/entropy using force constant matrix
Support calculation of phonon band with long-range Coulomb interaction considered
Support calculation of dielectric tensors using the linear response method
Support calculation of piezoelectric tensors using the linear response method
Support calculation of Born effective charges using the linear response method
Support Bader charge analysis
Support constraining lattice degrees of freedom along specified dimensions during structure optimization.
Feature Optimization
Supported .paw (Hongzhiwei PAW pseudopotential format) / .potcar (VASP POTCAR pseudopotential format) / .pawpsp (GBRV PAW pseudopotential format)
Added a preconditioned conjugate gradient method in the self-consistent iteration algorithm.
Added a fast inertial relaxation method in NEB relaxation.
Added a convergence criterion option for energy convergence in structure relaxation and NEB calculations.
Added support for modifying the Alpha and Omega coefficients in hybrid functionals, and accelerated hybrid functional calculations using the Adaptively Compressed Exchange Operator.
Added projected magnetic moment information, maximum force during structure relaxation, maximum force during transition state search, and band gap information in the output file.
Added a temporary calculation folder paw_tmp within the calculation directory to store intermediate files and error messages.
2021B
New Features
Support for CI-NEB method for transition state search
Supports hybrid functionals PBE0, HSE03, and HSE06.
Support DFT-D2 and DFT-D3 van der Waals corrections
Supports calculation of dielectric constant, refractive index, reflectivity, absorption coefficient, extinction coefficient, and more
Support calculations for charged systems.
Support spin-orbit coupling
Support phonon band structure and density of states calculations using the finite displacement method.
Support phonon band structure and density of states calculations using the DFPT method
Support DFT+U for strongly correlated systems
Support first-principles molecular dynamics calculations
2021beta
New Features
Using a plane-wave basis set to expand the wavefunctions
Using the Projector Augmented-Wave (PAW) method for pseudopotentials
Structure relaxation calculations, supporting atomic position relaxation, lattice relaxation, and lattice and atomic position relaxation
Self-consistent field (SCF) calculations
Support non-spin-polarized, collinear spin-polarized, non-collinear spin-polarized, and spin-orbit coupling systems
Total energy calculation
Atomic force calculation
Stress calculation
Band structure (projected band structure) calculation
Electronic density of states (projected density of states) calculation
Electron Localization Function (ELF) calculation
Potential calculation, supporting electrostatic potential and local potential calculations